Welcome to Chemistry in Solution and at Interfaces (CSI)
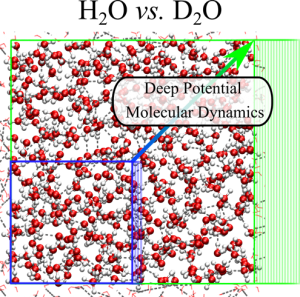
The green frames depict the snapshots of a deep potential molecular dynamics (DPMD) simulation of liquid water. The blue frames depict the snapshots of an ab-initio molecular dynamics (AIMD) simulation for the same system. The interatomic potential energy surface (PES) of AIMD is derived on the fly from the instantaneous ground-state of the electrons within density functional theory. In DPMD, the PES is represented by a deep-neural network that learned the surface dependence on the nuclear coordinates from ab-initio data using the software tools developed at CSI. DPMD extends significantly the accessible length and time scales of AIMD without losing accuracy and/or ability to model cleavage and formation of chemical/physical bonds. When used in conjunction with Feynman path integral techniques, DPMD enables extensive sampling of thermal and nuclear quantum fluctuations for systems in condensed-phase, such as neat water and water solutions in the bulk and in contact with material interfaces.
CSI is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences
 |
 |
![]()
![]()